RNA splicing is an essential, developmentally regulated biochemical reaction used by all known eukaryotes to “decode” their genomes. For incompletely understood reasons, genes are interrupted by “intron” sequences that need to be removed from the RNA so that the mature RNA can carry out its required functions as a protein-coding template or non-coding RNA. Eukaryotes excise introns by an ancient process thought to have arisen in the last eukaryotic common ancestor (LECA), and one with a multitude of important biomedical implications. RNAs can be spliced in alternative patterns, such that an RNA from a single gene can have opposite functions. Dysregulation of splicing causes human diseases and influences carcinogenesis and cancer progression. Despite the importance of splicing, the functions of all but a handful of the more than two million human RNA isoforms are unknown.
We use data-driven discovery, innovative experimental design, and biological validation to:
precisely detect spliced RNAs
determine how these spliced RNAs are regulated, and
understand how each RNA functions
Deciphering transcriptomic and genomic diversifying mechanisms
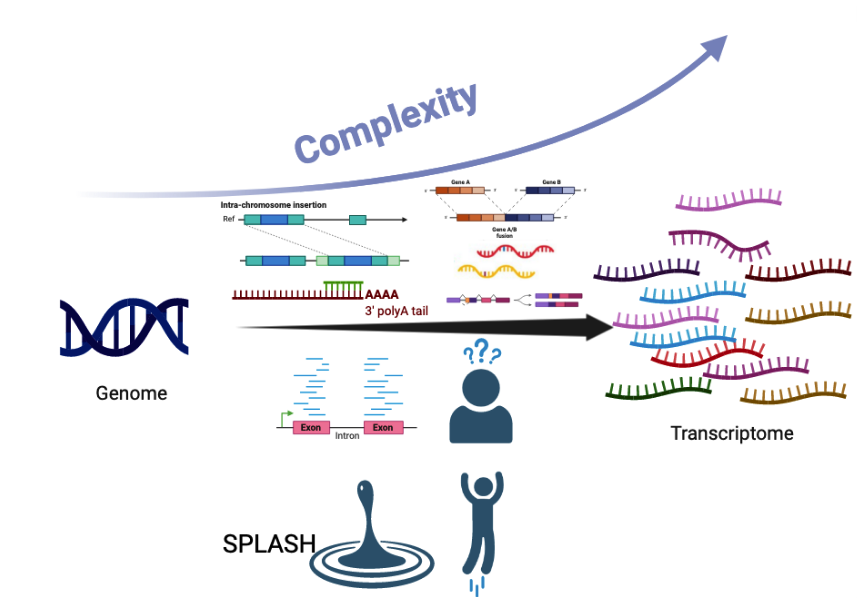
Myriad mechanisms diversify the sequence content of eukaryotic transcripts at the DNA and RNA levels with profound functional consequences. Examples include diversity generated by RNA splicing and V(D)J recombination. These and other events are detected with fragmented bioinformatic tools that require predefining a form of transcript diversification; moreover, they rely on alignment to a necessarily incomplete reference genome, filtering out unaligned sequences which can be among the most interesting. Each of these steps introduces blindspots for discovery. We have developed SPLASH, a new analytic method that performs unified, reference-free statistical inference directly on raw sequencing reads
Regulation of alternative splicing
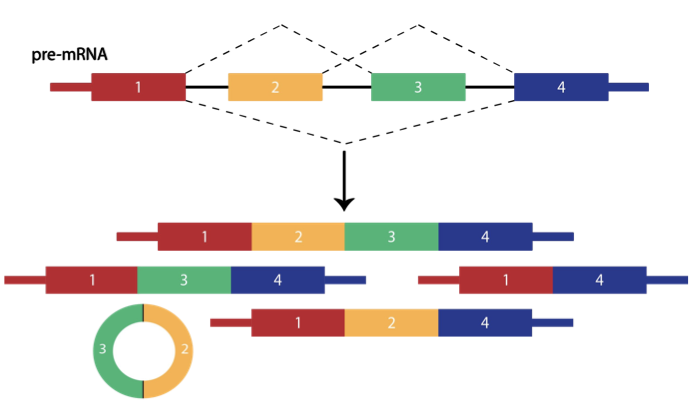
Alternative splicing of RNAs is an essential source of phenotypic variation between cell types, tissues, and organisms, and dysregulated alternative splicing can lead to disease. Despite the critical importance of alternative splicing in virtually all eukaryotes, it is currently impossible to predict which isoforms will be produced before a pre-mRNA is spliced. We are investigating the effects of cis-regulation, e.g. by repetitive elements, and trans-regulation, e.g. by non-coding RNAs, with the goal of building predictive statistical models of splicing.
Splicing in single cells
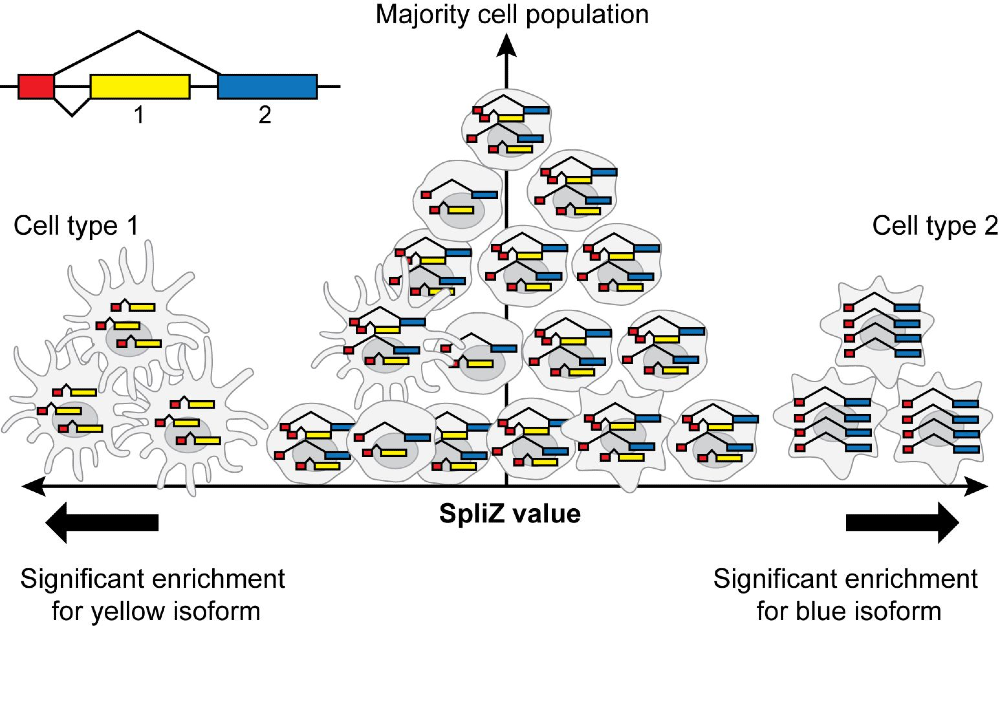
The recent boom in single-cell RNA sequencing has enabled widespread analysis at the gene level, but splicing in single cells is still rarely analyzed. We use rigorous statistical methods to deconvolve sources of noise in single-cell RNA-Seq data (Dehghannasiri et al, 2021), enabling us to filter out low-quality data at single-cell resolution. We have also developed the SpliZ, a rigorous method to uncover differential splicing and splicing regulation at the single-cell level (Olivieri et al, 2021; Olivieri et al, 2021).
Circular RNA
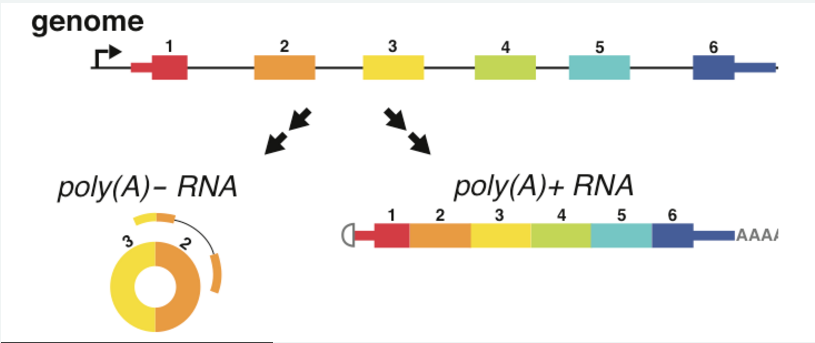
In 2012 Julia Salzman made the surprising discovery that circular RNA (circRNA) are a transcriptional product in thousands of eukaryotic genes (Salzman et al, 2012) and in hundreds of cases constitute the dominant RNA isoform. We developed the first methods to accurately detect circRNA in RNA-Seq data (Szabo et al, 2015). In addition, our lab continues to identify and study the mechanisms of circular RNA biogenesis (Barrett et al, 2015). We are currently developing methods to detect and analyze circRNA at the single-cell level.
Spatial RNA-Seq
A major limitation of single-cell RNA-Seq is that the spatial context of the cells is not maintained. Spatial RNA sequencing is a new field of transcriptomics that aims to determine both the location of cells in a sample and which RNAs they express. We are working to develop the next generation of spatial RNA sequencing simple enough to plug into existing single-cell RNA-Seq pipelines.
Gene fusions

Gene fusions are one of the hallmarks of cancer and are among the most powerful biomarkers and drug targets in translational cancer genomics. Massive public cancer sequencing databases provide a unique opportunity for detecting novel fusions via deploying computational algorithms. However, developing precise fusion detection algorithms is still a challenging problem due to the inordinate number of false positives. To overcome these challenges, our lab has developed highly specific statistical algorithms, DEEPEST and MACHETE (Dehghannasiri et al, 2019; Hseih et all, 2017), for fusion detection.
SQUICH
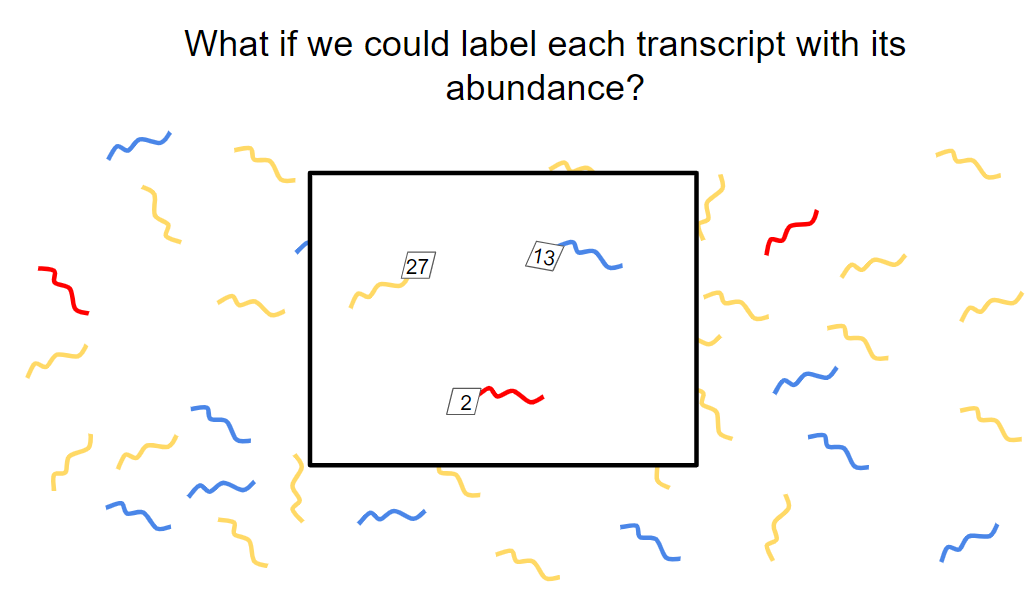
Current sequencing, even with depletion and enrichment schemes, still relies on simple random sampling of the remaining sequences. Therefore, a rare sequence that occurs only a few times might be missed, especially in cases where read depth is low. Depleting highly abundant sequences also results in information loss for those abundant transcripts.
What if we could somehow label a small subset of each transcript with its abundance and, by sequencing only this subset, deduce the original levels of each transcript? SQUICH (SeQUential depletIon and enriCHment) aims to do just that (Horn et al, 2019).
Get Involved
Please contact Julia (julia.salzman at stanford.edu) if you are interested in learning more about or becoming involved in our work which draws from
Statistics
Biochemistry
Genetics
Bioinformatics
Data Science
High school students interested in summer work should apply through the SIMR or Genecamp programs.