Software developed in the Salzman lab
We produce sensitive and specific tools for making discoveries in next generation sequencing data through the use of statistical modeling. Our tools are used by members of academia, medicine, and industry alike.
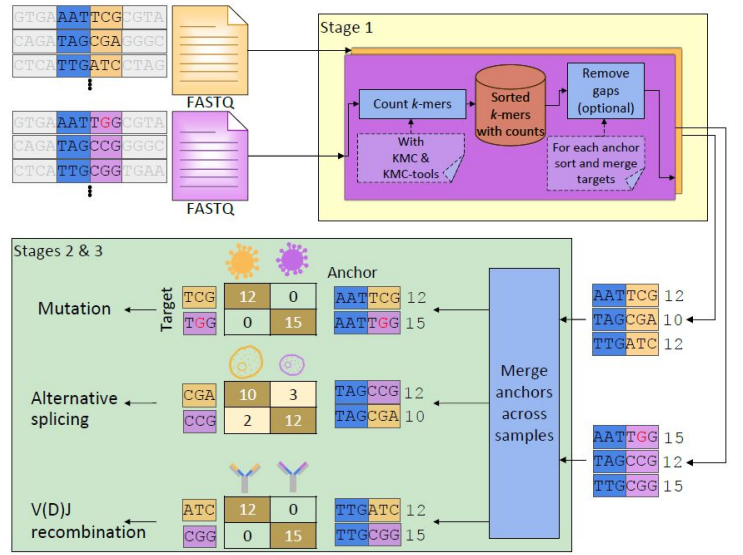
SPLASH2
SPLASH2 is an ultra-efficient and scalable software for unsupervised discovery directly on raw sequencing reads
(developed in collaboration with REFRESH Bioinformatics Group at Silesian University of Technology: https://refresh-bio.github.io/)
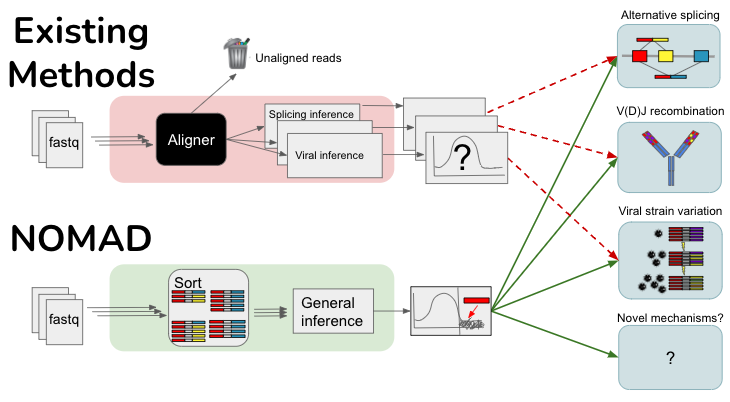
SPLASH
SPLASH is a reference-free, highly efficient algorithm to solve many fundamental problems in genome science.
Python version: https://github.com/salzman-lab/SPLASH
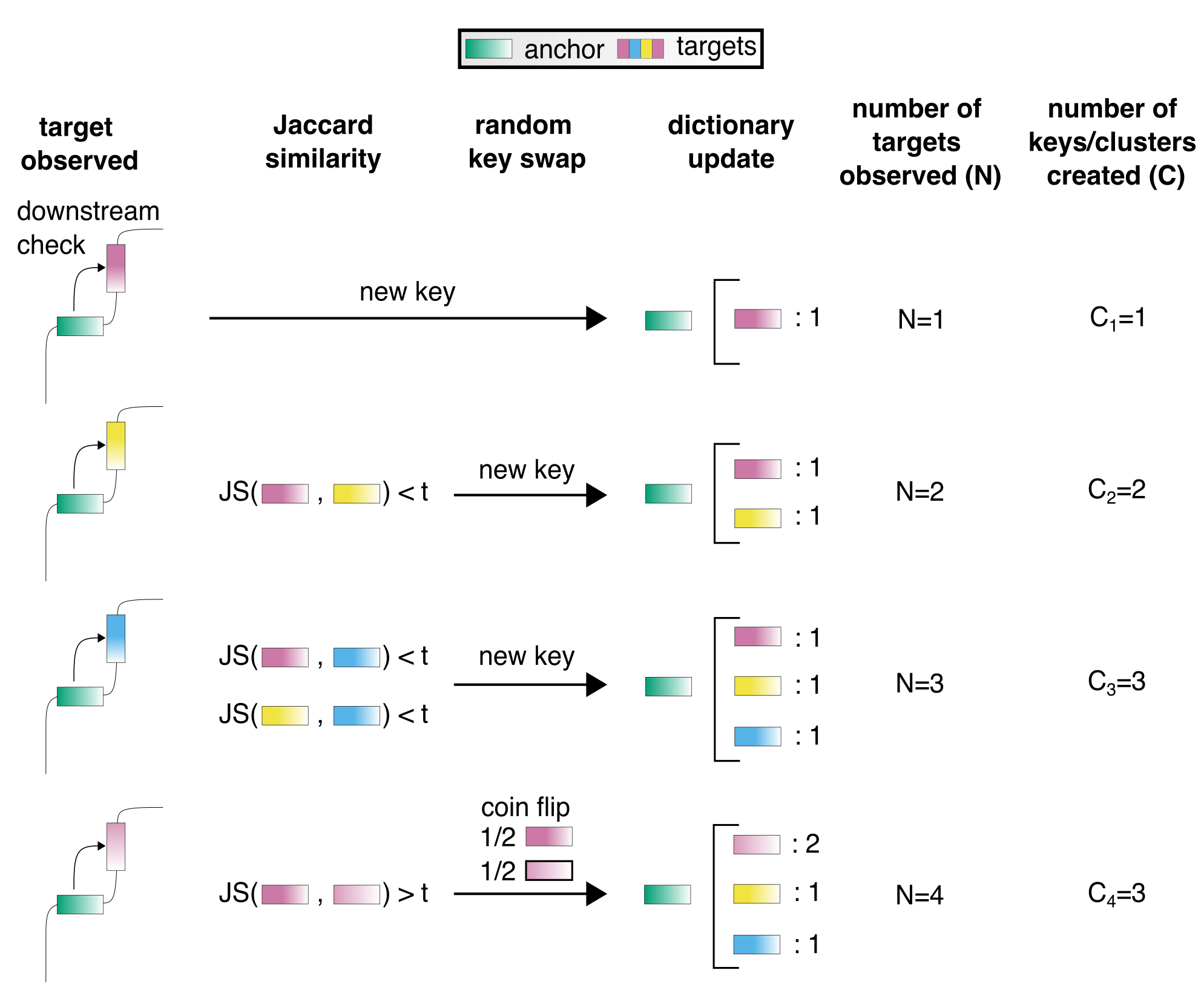
DIVE
DIVE is a reference-free statistical approach to diversity-generating and mobile genetic element discovery.
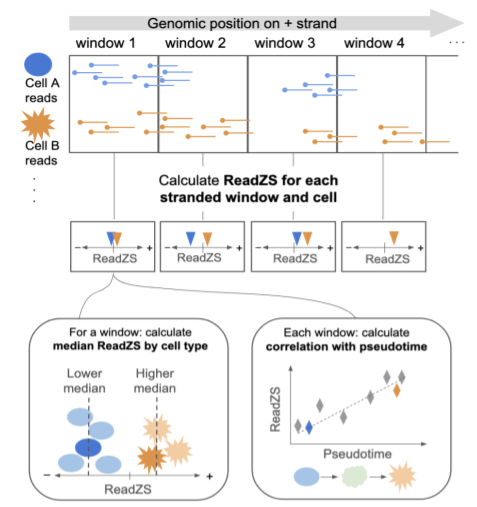
ReadZS
The ReadZS is a computationally-efficient, and annotation-free statistical approach to identify regulated RNAP, including but not limited to APA, in single cells. ReadZS rediscovers and substantially extends the scope of known cell type-specific RNAP in the human lung and during human spermatogenesis.
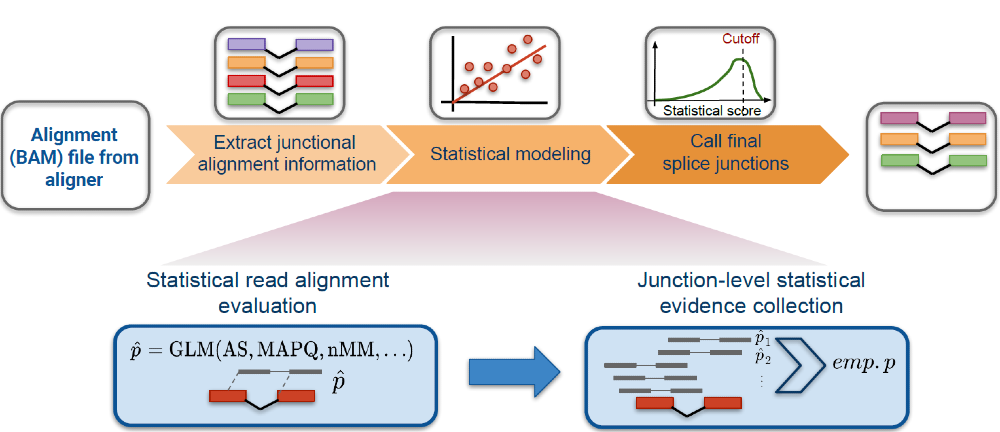
SICILIAN
SICILIAN is a statistical method for identifying RNA splice junctions using alignments reported from a spliced aligner. SICILIAN is currently implemented for the STAR aligner and will be adapted to more spliced aligners in the near future.
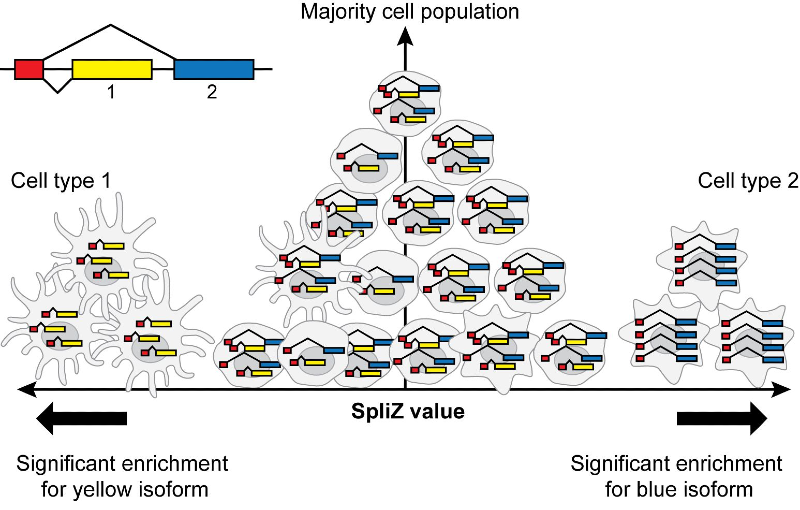
The SpliZ (Splicing Z score)
The SpliZ pipeline provides a novel, robust, and computationally efficient set of statistics to detect regulated splicing in single cell RNA-seq including 10x Chromium. The SpliZ(VD) provides annotation-free detection of differentially regulated, complex alternative splicing events.
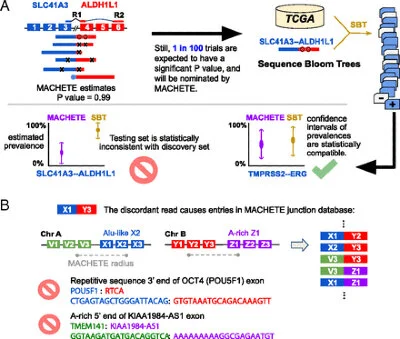
DEEPEST (Data-Enriched Efficient PrEcise STatistical fusion detection)
A highly specific and efficient statistical fusion detection pipeline specially designed for mining massive sequencing databases.
*There is also an online App with a web-based user interface for DEEPEST. The app can be run with only few clicks on a cloud environment (Cancer Genomics Cloud). Publication
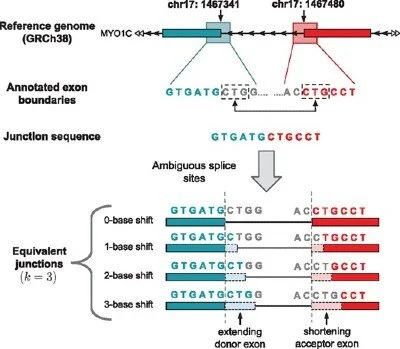
Equivalent Junctions
A method to detect the set of neighboring junctions that result in the same junction sequence.
Publication
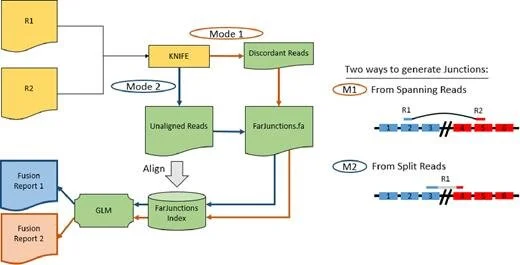
MACHETE (Mismatched Alignment CHimera Tracking Engine)
A statistical method to detect gene fusions at annotated splicing boundaries.
Publication
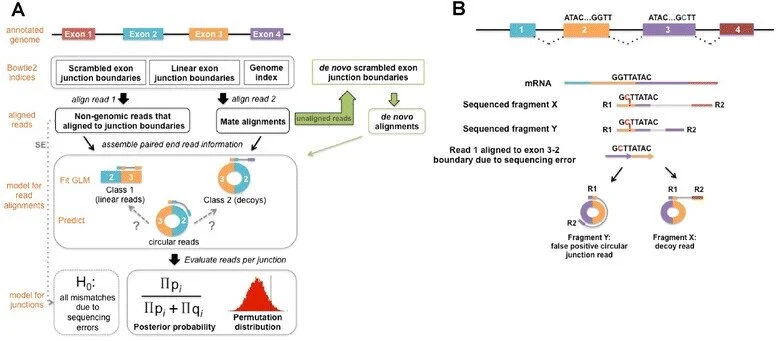
KNIFE (Known and Novel Isoform Explorer)
A method to detect circular RNA both at annotated and unannotated boundaries.
Publication